What is IPEX?
Overview
IPEX Syndrome, which stands for "Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked" syndrome, is a rare genetic disorder primarily affecting boys due to its X-linked recessive inheritance pattern.
It is characterized by severe autoimmune disorders that typically manifest during infancy or early childhood. The dysfunction arises from mutations in the FOXP3 gene, which is crucial for the development and regulation of T cells, particularly regulatory T cells that help prevent autoimmune reactions.
Individuals with IPEX Syndrome commonly exhibit a triad of symptoms: eczematous dermatitis (skin rashes), type 1 diabetes mellitus, and autoimmune enteropathy, which presents as persistent diarrhea. Other symptoms may include kidney dysfunction, thyroid disease, growth retardation, and susceptibility to infections, amongst others.
The condition can lead to life-threatening complications if not diagnosed and treated promptly, making early intervention critical.
Diagnosis typically involves a comprehensive evaluation of clinical symptoms, genetic testing to identify mutations in the FOXP3 gene, and additional tests to assess immune system functionality.
Treatment focuses on managing symptoms and may include immunosuppressive therapies, nutritional support, and in some cases, bone marrow transplantation to restore normal immune function. Supportive therapies such as antibiotics and antifungals are often necessary to prevent infections in affected individual.
Due to the complexity of the syndrome, a multidisciplinary approach involving various healthcare specialists is essential for managing the condition effectively and improving the quality of life for those affected.
Times are changing, however, and thanks to the work of many incredible people, a cure is on the horizon!

Genetics
The immune system uses a variety of mechanisms to restrain autoimmune reactions. This includes a group known as regulatory T cells, or Treg cells.
These Tregs play a powerful role in controlling or restraining the actions of other immune cells. In fact, they are like a small police force that has the job of monitoring other immune cells to make sure that they don’t respond too strongly, or inappropriately target other cells in the person's body.
One of the key proteins required for Tregs to develop and function normally is called FOXP3. In people with IPEX, the X-chromosome gene responsible for creating FOXP3 is faulty, resulting in an uncontrolled Treg and autoimmune response.
The FOXP3 gene lives on the X chromosome, and is recessive. This means that a mother with a faulty X chromosome has a 50% chance of passing it to their child. Sons with that X chromosome are likely to have IPEX, while daughters will be asymptomatic and carry a similar 50% risk of passing the faulty gene down.
History
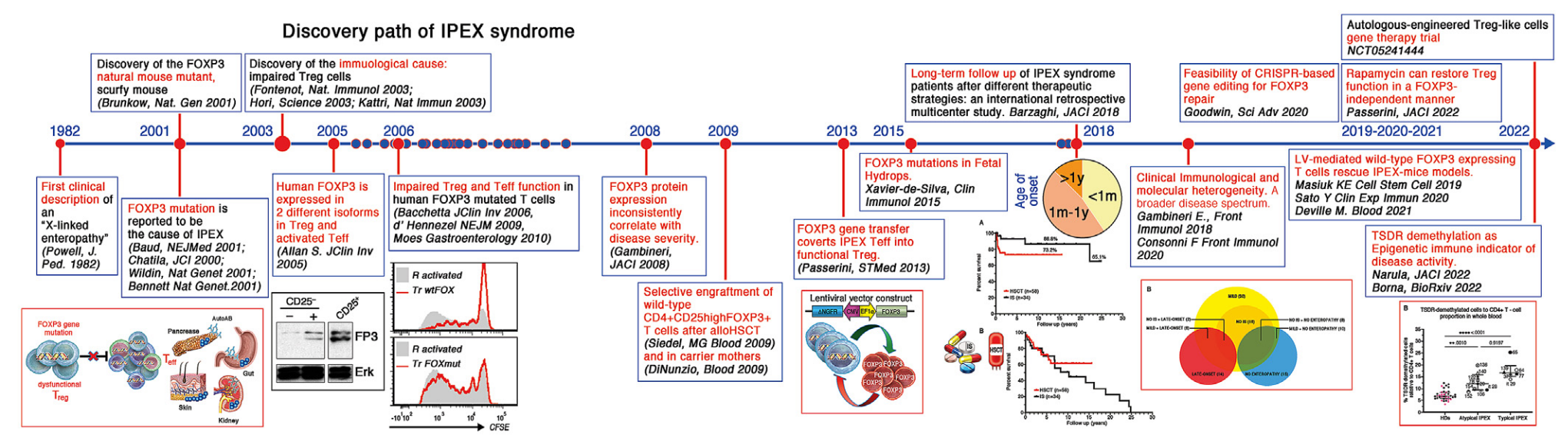
1982: First clinical description of "X-linked enteropathy" (Powell, J. Ped. 1982)
2001: Discovery of the FOXP3 natural mouse mutant, scurfy mouse (Brunkow, Nat. Gen 2001)
2001: FOXP3 mutation is reported to be the cause of IPEX (Baud, NEJMed 2001; Chatila, JCI 2000; Wildin, Nat Gen 2001; Bennett, Nat Gen 2001)
2003: Discovery of the immunological cause: impaired Treg cells (Fontenot, Nat Immunol 2003; Hori, Science 2003; Khattri, Nat Immun 2003)
2005: Human FOXP3 is expressed in 2 different isoforms in Treg and activated Teff (Allan S, JClin Inv 2005)
2006: Impaired Treg and Teff function in human FOXP3 mutated T cells (Bacchetta, JClin Inv 2006; d'Hennezel, NEJM 2009; Moes, Gastroenterology 2010)
2008: FOXP3 protein expression inconsistently correlate with disease severity (Gambineri, JACI 2008)
2009: Selective engraftment of wild-type CD4+CD25highFOXP3+T cells after alloHSCT (Seidel MG, Blood 2009) and in carrier mothers (Di Nunzio, Blood 2009)
2013: FOXP3 gene transfer converts IPEX Teff into functional Treg (Passerini, STMed 2013)
2015: FOXP3 mutations in Fetal Hydrops (Xavier-de-Silva, Clin Immunol 2015)
2018: Long-term follow up of IPEX syndrome patients after different therapeutic strategies: an international retrospective multicenter study (Barzaghi, JACI 2018)
2018: Clinical immunological and molecular heterogeneity. A broader disease spectrum (Gambineri E., Front Immunol 2018, Consonni F, Front Immunol 2021)
2019: LV-mediated wild-type FOXP3 expressing T cells rescue IPEX-mice model (Masiuk KE, Cell Stem Cell 2019, Sato Y., Clin Exp Immun 2020, Deville M., Blood 2021)
2020: Feasibility of CRISPR-based gene editing for FOXP3 repair (Goodwin, Sci Adv 2020)
2022: Rapamycin can restore Treg function in a FOXP3-independent manner (Passerini, JACI 2022)
2022: TSDR demethylation as Epigenetic immune indicator of disease activity (Narula, JACI 2023, Borna, STMed 2023)
2022-Ongoing: Autologous-engineered Treg-like cells gene therapy trial (NCT05241444)