Qu'est-ce que l'IPEX?
Aperçu
Le syndrome IPEX, qui signifie « syndrome de dysrégulation immunitaire, polyendocrinopathie, entéropathie, lié à l'X », est une maladie génétique rare qui touche principalement les garçons en raison de son mode de transmission récessif lié à l'X.
Elle se caractérise par des troubles auto-immuns graves qui se manifestent généralement au cours de la petite enfance. Le dysfonctionnement résulte de mutations du gène FOXP3, qui est essentiel au développement et à la régulation des lymphocytes T, en particulier des lymphocytes T régulateurs qui aident à prévenir les réactions auto-immunes.
Les personnes atteintes du syndrome IPEX présentent généralement une triade de symptômes : dermatite eczémateuse ( éruptions cutanées ), diabète sucré de type 1 et entéropathie auto-immune, qui se présente sous la forme d' une diarrhée persistante . D'autres symptômes peuvent inclure un dysfonctionnement rénal , une maladie thyroïdienne , un retard de croissance et une sensibilité aux infections , entre autres.
Cette maladie peut entraîner des complications potentiellement mortelles si elle n’est pas diagnostiquée et traitée rapidement, ce qui rend une intervention précoce essentielle.
Le diagnostic implique généralement une évaluation complète des symptômes cliniques, des tests génétiques pour identifier les mutations du gène FOXP3 et des tests supplémentaires pour évaluer la fonctionnalité du système immunitaire.
Le traitement se concentre sur la gestion des symptômes et peut inclure des thérapies immunosuppressives, un soutien nutritionnel et, dans certains cas, une greffe de moelle osseuse pour restaurer une fonction immunitaire normale. Des thérapies de soutien telles que des antibiotiques et des antifongiques sont souvent nécessaires pour prévenir les infections chez les personnes touchées.
En raison de la complexité du syndrome, une approche multidisciplinaire impliquant divers spécialistes de la santé est essentielle pour gérer efficacement la maladie et améliorer la qualité de vie des personnes touchées.
Mais les temps changent et grâce au travail de nombreuses personnes incroyables, un remède est à l’horizon !

Essais cliniques et actualités
Mises à jour sur les essais et les traitements pour les patients atteints d'IPEX
Génétique
Le système immunitaire utilise divers mécanismes pour limiter les réactions auto-immunes. Cela inclut un groupe connu sous le nom de cellules T régulatrices, ou cellules Treg.
Ces Tregs jouent un rôle important dans le contrôle ou la limitation des actions d'autres cellules immunitaires. En fait, ils sont comme une petite force de police chargée de surveiller les autres cellules immunitaires pour s'assurer qu'elles ne réagissent pas trop fortement ou ne ciblent pas de manière inappropriée d'autres cellules du corps de la personne.
L'une des protéines clés nécessaires au développement et au fonctionnement normal des Tregs s'appelle FOXP3. Chez les personnes atteintes d'IPEX, le gène du chromosome X responsable de la création de FOXP3 est défectueux, ce qui entraîne une réponse Treg incontrôlée et auto-immune.
Le gène FOXP3 est présent sur le chromosome X et est récessif. Cela signifie qu'une mère présentant un chromosome X défectueux a 50 % de chances de le transmettre à son enfant. Les garçons porteurs de ce chromosome X sont susceptibles d'être atteints d'IPEX, tandis que les filles seront asymptomatiques et auront un risque similaire de 50 % de transmettre le gène défectueux.
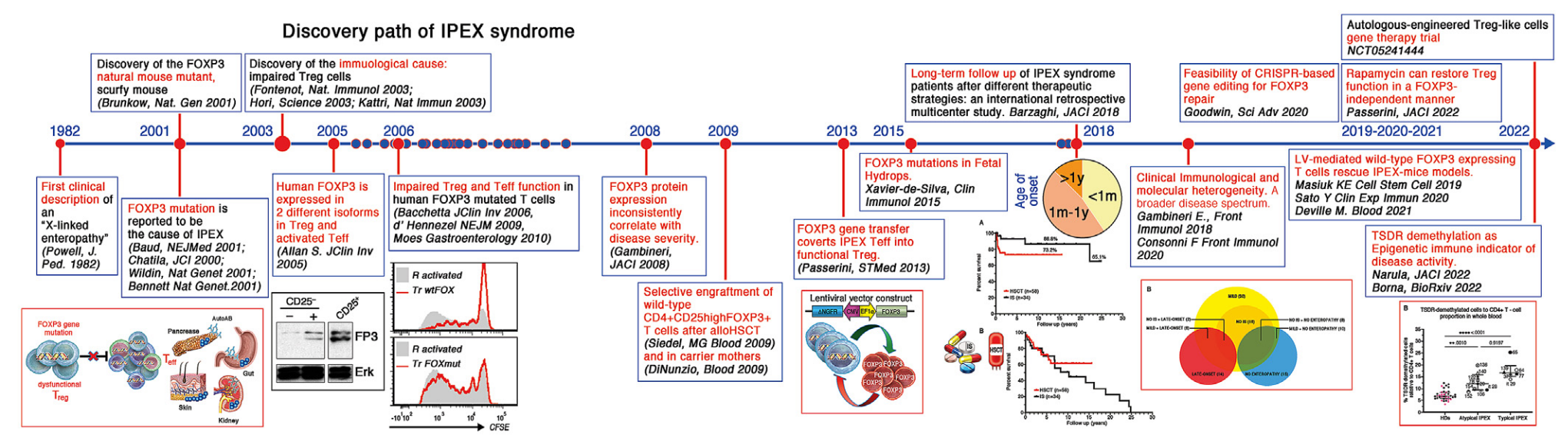
Histoire
1982 : Première description clinique de « l'entéropathie liée au chromosome X » ( Powell, J. Ped. 1982 )
2001 : Découverte du mutant naturel de souris FOXP3, la souris scurfy ( Brunkow, Nat. Gen 2001 )
2001 : La mutation FOXP3 serait la cause de l'IPEX (Baud, NEJMed 2001 ; Chatila, JCI 2000 ; Wildin, Nat Gen 2001 ; Bennett, Nat Gen 2001 )
2003 : Découverte de la cause immunologique : des cellules Treg altérées ( Fontenot, Nat Immunol 2003 ; Hori, Science 2003 ; Khattri, Nat Immun 2003 )
2005 : Le FOXP3 humain est exprimé dans 2 isoformes différentes dans les Treg et les Teff activés ( Allan S, JClin Inv 2005 )
2006 : Altération de la fonction Treg et Teff dans les cellules T mutées FOXP3 humaines ( Bacchetta, JClin Inv 2006 ; d'Hennezel, NEJM 2009 ; Moes, Gastroenterology 2010)
2008 : l'expression de la protéine FOXP3 est incohérente avec la gravité de la maladie ( Gambineri, JACI 2008 )
2009 : Greffe sélective de cellules T CD4+CD25highFOXP3+ de type sauvage après alloHSCT ( Seidel MG, Blood 2009 ) et chez des mères porteuses ( Di Nunzio, Blood 2009 )
2013 : Le transfert du gène FOXP3 convertit IPEX Teff en Treg fonctionnel ( Passerini, STMed 2013 )
2015 : Mutations du gène FOXP3 dans l'hydropisie fœtale (Xavier-de-Silva, Clin Immunol 2015)
2018 : Suivi à long terme des patients atteints du syndrome IPEX après différentes stratégies thérapeutiques : une étude multicentrique rétrospective internationale ( Barzaghi, JACI 2018 )
2018 : Hétérogénéité clinique, immunologique et moléculaire. Un spectre de maladies plus large ( Gambineri E., Front Immunol 2018 , Consonni F, Front Immunol 2021 )
2019 : Les lymphocytes T exprimant FOXP3 de type sauvage médiés par LV sauvent le modèle de souris IPEX ( Masiuk KE, Cell Stem Cell 2019 , Sato Y., Clin Exp Immun 2020 , Deville M., Blood 2021)
2020 : Faisabilité de l'édition génétique basée sur CRISPR pour la réparation de FOXP3 ( Goodwin, Sci Adv 2020 )
2022 : La rapamycine peut restaurer la fonction Treg de manière indépendante de FOXP3 ( Passerini, JACI 2022 )
2022 : La déméthylation du TSDR comme indicateur immunitaire épigénétique de l'activité de la maladie ( Narula, JACI 2023 , Borna, STMed 2023 )
2022-En cours : Essai de thérapie génique à base de cellules Treg autologues modifiées ( NCT05241444 )