¿Qué es IPEX?
Descripción general
El síndrome IPEX, que significa "desregulación inmunitaria, poliendocrinopatía, enteropatía, ligado al cromosoma X", es un trastorno genético poco común que afecta principalmente a los niños debido a su patrón de herencia recesivo ligado al cromosoma X.
Se caracteriza por trastornos autoinmunes graves que se manifiestan típicamente durante la infancia o la niñez temprana. La disfunción surge de mutaciones en el gen FOXP3, que es crucial para el desarrollo y la regulación de las células T, en particular las células T reguladoras que ayudan a prevenir las reacciones autoinmunes.
Las personas con síndrome IPEX suelen presentar una tríada de síntomas: dermatitis eccematosa ( erupciones cutáneas ), diabetes mellitus tipo 1 y enteropatía autoinmune, que se presenta como diarrea persistente . Otros síntomas pueden incluir disfunción renal , enfermedad tiroidea , retraso del crecimiento y susceptibilidad a infecciones , entre otros.
La afección puede provocar complicaciones potencialmente mortales si no se diagnostica y trata rápidamente, por lo que la intervención temprana es fundamental.
El diagnóstico generalmente implica una evaluación integral de los síntomas clínicos, pruebas genéticas para identificar mutaciones en el gen FOXP3 y pruebas adicionales para evaluar la funcionalidad del sistema inmunológico.
El tratamiento se centra en controlar los síntomas y puede incluir terapias inmunosupresoras, apoyo nutricional y, en algunos casos, trasplante de médula ósea para restablecer la función inmunitaria normal. A menudo, son necesarias terapias de apoyo, como antibióticos y antimicóticos, para prevenir infecciones en las personas afectadas.
Debido a la complejidad del síndrome, un enfoque multidisciplinario que involucre a varios especialistas de la salud es esencial para manejar la condición de manera efectiva y mejorar la calidad de vida de los afectados.
Sin embargo, los tiempos están cambiando y, gracias al trabajo de muchas personas increíbles, ¡la cura está a la vuelta de la esquina!

Ensayos clínicos y novedades
Actualizaciones sobre ensayos y tratamientos para pacientes con IPEX
Genética
El sistema inmunitario utiliza diversos mecanismos para frenar las reacciones autoinmunes, entre los que se incluye un grupo conocido como células T reguladoras o células Treg.
Estas células Treg desempeñan un papel importante en el control o la restricción de las acciones de otras células inmunitarias. De hecho, son como una pequeña fuerza policial que tiene la tarea de supervisar a otras células inmunitarias para asegurarse de que no respondan con demasiada fuerza o ataquen de forma inapropiada a otras células del cuerpo de la persona.
Una de las proteínas claves necesarias para que las células Treg se desarrollen y funcionen normalmente se llama FOXP3. En las personas con IPEX, el gen del cromosoma X responsable de crear FOXP3 es defectuoso, lo que da como resultado una respuesta autoinmune y descontrolada de las células Treg.
El gen FOXP3 se encuentra en el cromosoma X y es recesivo. Esto significa que una madre con un cromosoma X defectuoso tiene un 50 % de probabilidades de transmitirlo a su hijo. Los hijos varones con ese cromosoma X tienen más probabilidades de tener IPEX, mientras que las hijas serán asintomáticas y tendrán un riesgo similar del 50 % de transmitir el gen defectuoso a sus descendientes.
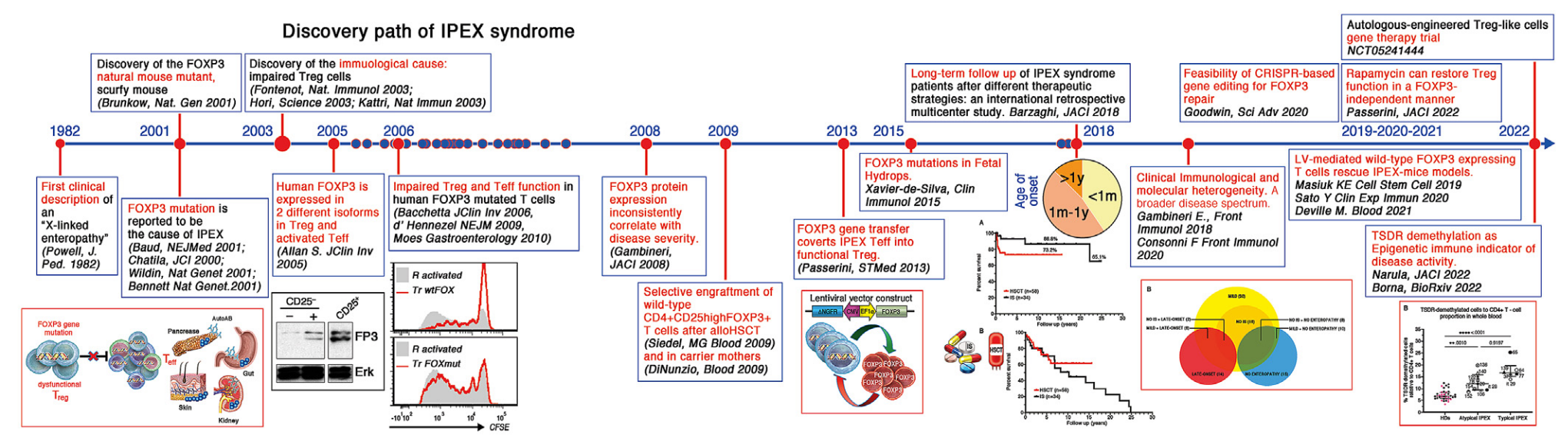
Historia
1982: Primera descripción clínica de la "enteropatía ligada al cromosoma X" ( Powell, J. Ped. 1982 )
2001: Descubrimiento del mutante natural FOXP3, el ratón casposo ( Brunkow, Nat. Gen 2001 )
2001: Se informa que la mutación FOXP3 es la causa de IPEX (Baud, NEJMed 2001; Chatila, JCI 2000 ; Wildin, Nat Gen 2001 ; Bennett, Nat Gen 2001 )
2003: Descubrimiento de la causa inmunológica: células Treg deterioradas ( Fontenot, Nat Immunol 2003 ; Hori, Science 2003 ; Khattri, Nat Immun 2003 )
2005: El FOXP3 humano se expresa en 2 isoformas diferentes en Treg y Teff activado ( Allan S, JClin Inv 2005 )
2006: Deterioro de la función de Treg y Teff en células T humanas con mutación FOXP3 ( Bacchetta, JClin Inv 2006 ; d'Hennezel, NEJM 2009 ; Moes, Gastroenterology 2010)
2008: La expresión de la proteína FOXP3 se correlaciona de manera inconsistente con la gravedad de la enfermedad ( Gambineri, JACI 2008 )
2009: Injerto selectivo de células T CD4+CD25highFOXP3+ de tipo salvaje después de un trasplante aloHSCT ( Seidel MG, Blood 2009 ) y en madres portadoras ( Di Nunzio, Blood 2009 )
2013: La transferencia del gen FOXP3 convierte el IPEX Teff en Treg funcionales ( Passerini, STMed 2013 )
2015: Mutaciones de FOXP3 en hidropesía fetal (Xavier-de-Silva, Clin Immunol 2015)
2018: Seguimiento a largo plazo de pacientes con síndrome IPEX después de diferentes estrategias terapéuticas: un estudio multicéntrico retrospectivo internacional ( Barzaghi, JACI 2018 )
2018: Heterogeneidad molecular e inmunológica clínica. Un espectro de enfermedades más amplio ( Gambineri E., Front Immunol 2018 , Consonni F, Front Immunol 2021 )
2019: Las células T que expresan FOXP3 de tipo salvaje mediadas por LV rescatan el modelo de ratones IPEX ( Masiuk KE, Cell Stem Cell 2019 , Sato Y., Clin Exp Immun 2020 , Deville M., Blood 2021)
2020: Viabilidad de la edición genética basada en CRISPR para la reparación de FOXP3 ( Goodwin, Sci Adv 2020 )
2022: La rapamicina puede restaurar la función de los Treg de manera independiente de FOXP3 ( Passerini, JACI 2022 )
2022: Desmetilación de TSDR como indicador inmunológico epigenético de la actividad de la enfermedad ( Narula, JACI 2023 , Borna, STMed 2023 )
2022-En curso: ensayo de terapia génica con células Treg similares a las autólogas ( NCT05241444 )